MKSAP Quiz: Follow-up evaluation of chronic anemia
A 32-year-old woman is seen for follow-up evaluation of chronic anemia that has been stable for at least the past 5 years. She has no history of transfusions. She had one pregnancy at age 29 years, ending in intrauterine fetal demise in the early third trimester. Her father has anemia. The patient is Chinese.
On physical examination, vital signs and the remainder of the physical examination are normal.
Laboratory studies:
| Hemoglobin | 10.8 g/dL (108 g/L) |
| Mean corpuscular volume | 62 fL |
| Reticulocyte count | 2% of erythrocytes |
| Iron studies | |
| Ferritin | 200 ng/mL (200 μg/L) |
| Iron | 200 μg/dL (36 μmol/L) |
| Total iron-binding capacity | 280 μg/dL (50 μmol/L) |
Hemoglobin electrophoresis reveals a normal migration pattern of hemoglobin A and normal hemoglobin A2 and hemoglobin F levels.
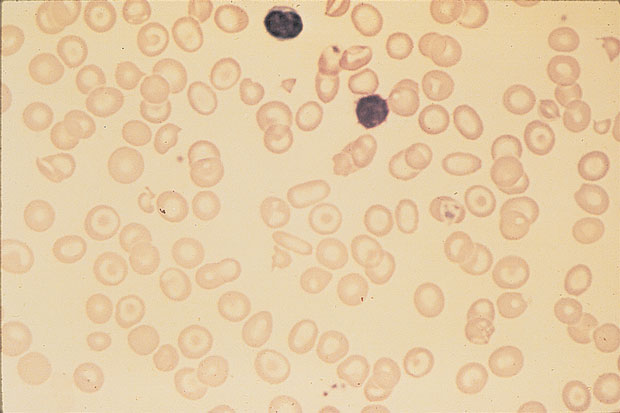
Peripheral blood smear is shown.
Which of the following is the most likely diagnosis?
A. α-Thalassemia carrier
B. α-Thalassemia trait
C. β-Thalassemia minor
D. Hemoglobin E trait
Answer and critique
This content is available to MKSAP 19 subscribers as Question 17 in the Hematology section.
The most likely diagnosis is α-thalassemia trait (Option B). This patient has a chronic hypochromic and microcytic anemia, with the presence of target cells on the peripheral blood smear characteristic of thalassemia. Patients with α-thalassemia trait have deletions of two or more of the four α-globin genes and present with a mild, microcytic and hypochromic anemia (hemoglobin level approximately 10 g/dL [100 g/L]) and a normal or elevated iron level. Thalassemia is common in African and Mediterranean countries, the Middle East, and Southeast Asia. Patients of Asian descent with α-thalassemia trait are more likely to have offspring with severe manifestations such as hemoglobin H (β-chain tetramers) or hydrops fetalis with hemoglobin Barts (intrauterine fetal demise resulting from deletion of all four α-chain genes). Patients with α-thalassemia trait will have mild anemia, but hemoglobin electrophoresis will be otherwise normal.
α-Thalassemia carrier (silent carrier) is defined as deletion of a single α-globin gene (−α/αα) (Option A). Silent carriers do not have anemia, and the mean corpuscular volume may be normal or only mildly decreased.
Patients with β-thalassemia have mutations of one copy or both copies of the β-globin gene leading to a reduction (β+) or complete absence (β°) of β-globin production (Option C). A reduced hemoglobin A level and elevated levels of minor hemoglobin components, hemoglobin F (α2/γ2), and hemoglobin A2 (α2/δ2) are seen on hemoglobin electrophoresis. This patient's normal hemoglobin electrophoresis is consistent with α-thalassemia rather than β-thalassemia.
Hemoglobin E is commonly found in Southeast Asia and results from a mutation in the β-globin gene (Option D). This mutation is associated with reduced expression of the β-globin gene and results in a β+ thalassemia minor phenotype (microcytosis, hypochromia, little or no anemia). A patient with hemoglobin E trait (heterozygous) would have microcytic and hypochromic erythrocytes, as in this patient, but the hemoglobin electrophoresis would show the presence of hemoglobin E (typically constituting less than 50% of the total hemoglobin).
Key Points
- Patients with α-thalassemia have chronic microcytic anemia, target cells on the peripheral blood smear, and a normal hemoglobin electrophoresis.
- Patients with β-thalassemia have chronic microcytic anemia, target cells on the peripheral blood smear, a reduced hemoglobin A level, and elevated levels of hemoglobin F and hemoglobin A2 on hemoglobin electrophoresis.