MKSAP Quiz: 1-year history of increasing fatigue
A 77-year-old man has a 1-year history of increasing fatigue. He has not seen a physician for at least 3 years. Medical and family histories are unremarkable, and he takes no medications.
On physical examination, the patient appears pale. Temperature is normal, blood pressure is 140/85 mm Hg, pulse rate is 72/min, and respiration rate is 16/min. There is no scleral icterus or jugular venous distention. Cardiopulmonary examination is normal. The spleen is palpable 6 cm below the left costal margin.
Laboratory studies:
| Hemoglobin: | 7.6 g/dL (76 g/L) |
| Leukocyte count: | 11,200/µL (11.2 × 109/L) |
| Platelet count: | 104,000/µL (104 × 109/L) |
| Haptoglobin: | Elevated |
| Direct antiglobulin (Coombs) test: | Negative |
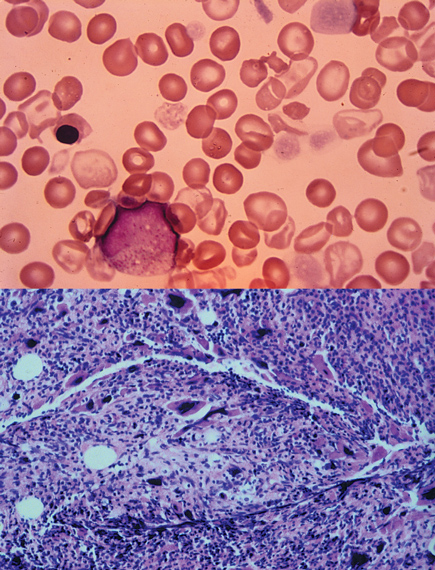
A bone marrow aspiration reveals a “dry tap.” A peripheral blood smear and bone marrow biopsy are shown.
Cytogenetic studies are negative for the Philadelphia chromosome.
Which of the following is the most likely diagnosis?
A. Aplastic anemia
B. Chronic myeloid anemia
C. Myelofibrosis
D. Pure red cell aplasia
Answer and critique
The correct answer is C: Myelofibrosis. This question can be found in MKSAP 15 in the Hematology and Oncology section, item 27.
Myeloproliferative syndromes are clonal stem cell disorders characterized by chronic unregulated hyperproliferation of the myeloid elements in the bone marrow (granulocytes, erythrocytes, platelets, and stromal cells). This patient has myelofibrosis, one of the four entities that constitute the myeloproliferative syndrome. Myelofibrosis causes clonal proliferation of abnormal stromal cells that release fibrosis-promoting cytokines in the bone marrow. His peripheral blood smear shows a classic leukoerythroblastic picture of immature granulocytes (a metamyelocyte) and early erythrocyte forms (nucleated erythrocytes) as well as dysmorphic erythrocytes (tear-drop cells). Splenomegaly and hepatomegaly result from extramedullary hematopoiesis, and patients can develop portal hypertension. Bone marrow hypercellularity is the hallmark of this disease and often results in dry bone marrow aspiration. This patient's bone marrow biopsy reveals typical findings of myelodysplasia, including a hypercellular marrow with dysmorphic megakaryocytes and no evidence of increased blast forms, resulting in inadequate hematopoiesis. Median survival is 3 to 5 years, and death commonly results from marrow failure, transformation to acute leukemia, or complications of portal hypertension.
Patients with chronic myeloid leukemia characteristically have an elevated leukocyte count, a low platelet count, and splenomegaly, but the leukocyte count is often higher than this patient's count, and the peripheral blood smear shows the characteristic full array of maturing granulocytes. Bone marrow is hypercellular with marked myeloid hyperplasia and minimal fibrosis, and cytogenetic analysis shows the Philadelphia chromosome.
Aplastic anemia is a disorder in which myeloid progenitor cells and stem cells are severely diminished or absent in the bone marrow because of an intrinsic defect of the stem cells or immune-mediated stem cell destruction, which leads to transfusion-dependent anemia, thrombocytopenia, and severe neutropenia. The bone marrow is typically aplastic or hypoplastic, without evidence of infiltrating malignant cells, and there is no evidence of hepatomegaly or splenomegaly.
Acquired chronic pure red cell aplasia is characterized by the absence or marked decrease of erythrocyte production, probably as the result of immune-mediated destruction of erythroid progenitors. Affected patients typically have severe normochromic and normocytic or macrocytic anemia as well as reticulocytopenia with normal leukocyte and platelet counts. Nucleated and dysmorphic erythrocytes are not present in the peripheral blood smear, and the bone marrow shows profound erythroid hypoplasia.
Key Point
- Myelofibrosis is characterized by splenomegaly, normocytic anemia, circulating erythroblasts and myeloid precursors, teardrop cells, and bone marrow fibrosis.